Dimerization And Activation
Definition noun, plural: dimerizations (1) The chemical reaction that joins two molecular subunits, resulting in the formation of a single dimer. (2) The process or act of forming a dimer. Supplement Dimerization may be heterodimerization or homodimerization depending on the nature of the subunits joined together. Unprecedented synthetic routes to various PAHs from simple phenol derivatives by a palladium‐catalyzed annulative dimerization of phenylene triflate through twofold inter‐ and intramolecular C−H activation have been established.
The objective of our research was to study the mechanisms of activation of mAb against the gp130 transducer chain common to the IL-6 cytokine family. It has been found that among the 56 anti-gp130 available worldwide, none was able to activate the growth of IL-6-dependent myeloma cell lines. When certain of them were associated in pairs they allowed the cells to grow; alone, they were inhibitory. The same activation was also obtained by cross-linking certain anti-gp130 mAb on the cell membrane with a goat anti-mouse Ig antiserum. A bispecific mAb was prepared by the somatic fusion of two hybridomas secreting two mAb whose association was able to activate gp130 signaling; the bispecific mAb was inactive. The activating mAb were able to support long-term proliferation of the IL-6-dependent myeloma cell lines, which indicates that they are potential valuable growth factors of tumor cells and hematopoietic stem cells. When they were injected into SCID mice, they allowed human IL-6-dependent myeloma cell lines to grow, develop tumors and metastasize. By studying the functional epitopes of the cell membrane gp130 receptors, it was shown that the activating mAb induced gp130 dimerization and STAT3 activation, as did IL-6.
Autophosphorylation is a type of post-translational modification of proteins. It is generally defined as the phosphorylation of the kinase by itself. In eukaryotes, this process occurs by the addition of a phosphate group to serine, threonine or tyrosine residues within protein kinases, normally to regulate the catalytic activity.[3][4] Autophosphorylation may occur when a kinases' own active site catalyzes the phosphorylation reaction (cis autophosphorylation), or when another kinase of the same type provides the active site that carries out the chemistry (trans autophosphorylation). The latter often occurs when kinase molecules dimerize.[3] In general, the phosphate groups introduced are gamma phosphates from nucleoside triphosphates, most commonly ATP.[3]
Function[edit]
Protein kinases, many of which are regulated by autophosphorylation, are vital in controlling the cellular proliferation, differentiation, metabolism, migration and survival. Mutations in the genes encoding them or their potential activators or repressors can affect any number of functions within an organism.[3][4] Phosphorylation is easily reversed by phosphatases. Therefore, it is an effective method of turning 'on' and 'off' kinase activity. Because of this it is recognized as an essential process in cell signaling.[3] Addition of a negatively charged phosphate group brings about a change in the microenvironment that may lead to attraction or repulsion of other residues or molecules.[3][4] The result may be a conformational change to expose or hide catalytic or allosteric seats from the surface.[3] If the phosphorylated residue resides within the catalytic seat itself, it may facilitate or prevent substrate binding by means of charge-interaction, or by providing or preventing complementary shapes necessary for molecular recognition.[3] In addition, the phosphate group yields several potential areas for hydrogen-bonding or establishment of salt-bridges, of which the latter generally involves an arginine residue.[3][5]
Binding of effector molecules may be affected in a similar manner if the phosphorylated residue makes part of the allosteric site.[3] Autophosphorylation has also been reported to have an effect on the cell's ability for endocytosis and proteolysis.[5]
Process and structure[edit]
Kinases are either phosphorylated on serine and/or threonine residues, or solely on tyrosine residues.[5] This serves as a means to classify them as either Ser/Thr- or Tyr-kinases. Several residues within the primary structure may be autophosphorylated simultaneously. The phosphoacceptors often reside within loops in the protein structure suitably termed 'activation loops'.[3] The structures of some autophosphorylation complexes are known from crystals of protein kinases in which the phosphorylation site (Ser, Thr, or Tyr) of one monomer in the crystal is sitting in the active site of another monomer of the crystal in a manner similar to known peptide-substrate/kinase structures.[6] The known structures include:
- Tyr phosphorylation sites in juxtamembrane regions:
- human cKIT, Tyr568 (PDB: 1PKG)[7]
- human CSF1R, Tyr561 (PDB: 3LCD, homologous to cKIT site)[6][8]
- human EPHA2, Tyr594 (PDB: 4PDO, two residues after the cKIT and CSF1R sites)[6]
- Tyr phosphorylation sites in kinase insert regions:
- human FGFR1, Tyr583 (PDB: 3GQI)[9]
- human FGFR3, Tyr577 (PDB: 4K33, homologous to the FGFR1 site,[6] domain interface identical to FGFR1 structure)[10]
- Tyr phosphorylation sites in activation loops:
- human IGF1R, Tyr1165 (PDB: 3D94)[11]
- human IGF1R, Tyr1166 (PDB: 3LVP)[6][12]
- human LCK, Tyr394 (PDB: 2PL0, homologous to the IGF1R Tyr1165 site[6])[6][13]
- Ser/Thr phosphorylation sites in activation loops:
- human PAK1, Thr423 (PDB: 3Q4Z, 4O0R, 4O0T, 4P90, 4ZLO, 4ZY4, 4ZY5, 4ZY6, 5DEY; the 4ZY4 and 4ZY5 structures provide complete coordinates for the substrate activation loop[6])[6][14][15][16][17]
- human IRAK4, Thr345 (PDB: 4U97, 4U9A)[18]
- N or C terminal tails Ser/Thr phosphorylation sites:
- C. elegans CaMKII, C-terminal tail, Thr284 (PDB: 3KK8, 3KK9)[19]
- human CaMKII, C-terminal tail, Thr287 (PDB: 2WEL, homologous to the C. elegans site)[20]
- human CLK2, N-terminal tail, Ser142 (PDB: 3NR9)[6]
In general, the structures of the phosphorylation of internal loops involve important domain-domain contacts that have been confirmed by site-directed mutagenesis, while the phosphorylation of positions in the N or C terminal tails more than 10 amino acids away from the kinase domain do not involve important domain-domain contacts away from the substrate binding site.[6]
Signaling pathways and trans-autophosphorylation[edit]
Among a number of various molecules, Receptor Tyrosine Kinases (RTKs ) play a critical role in transducing signals through a range of signaling pathways. All RTKs consists of an extracellular ligand binding region, a single transmembrane helix and a cytoplasmic region (the tyrosine kinase domain). Prior to ligand stimulation most RTKs present as a monomer on the surface of cells. Ligand binding to the extracellular domain induces dimerization. Dimerization of RTKs leads to autophosphorylation of tyrosine in the catalytic core of the dimer, and finally stimulation of the tyrosine kinase activity and cell signaling.[21] It is thus an example of a trans-autophosphorylation reaction, where one receptor subunit of the dimer phosphorylates the other subunit.[22]
Examples of RTKs which undergo autophosphorylation[edit]
Epidermal growth factor receptor[edit]
An example of RTKs that undergo autophosphorylation is the Epidermal Growth Factor receptor (EGFR ). EGFR was the first discovered example of RTKs. Following ligand binding, a conformational change occurs in the EGFR monomers. This leads to EGFR dimerization.[21] Dimerization brings the two receptors into close proximity. This stimulates the kinase activity of EGFR, which leads to transautophosphorylation on multiple tyrosine residues in C-terminal end of the molecule. The phosphorylated tyrosine residue can then serve as a docking site for downstream signaling proteins.[21] (Fig. 1).
Insulin receptors[edit]
Splash screen while gnu radio is loading issue 5 cfriedt. Another example is the binding of insulin to insulin receptors. Once released into the bloodstream insulin can bind to receptors on the surface of cells in muscle or other tissues. This receptor is a protein with an (αβ)2 quaternary structure. The two large α-subunits are extracellular, while the smaller β-subunits have a transmembrane domain as well as extra-and intracellular domains. In the absence of insulin, the two intracellular domains of the β subunits are separated. Binding with insulin triggers a conformational change in the receptor that brings them closer together (dimerization). Each β subunit intracellular domain is a tyrosine kinase that phosphorylates its partner in the receptor.[3]
Cancer[edit]
Src kinases[edit]
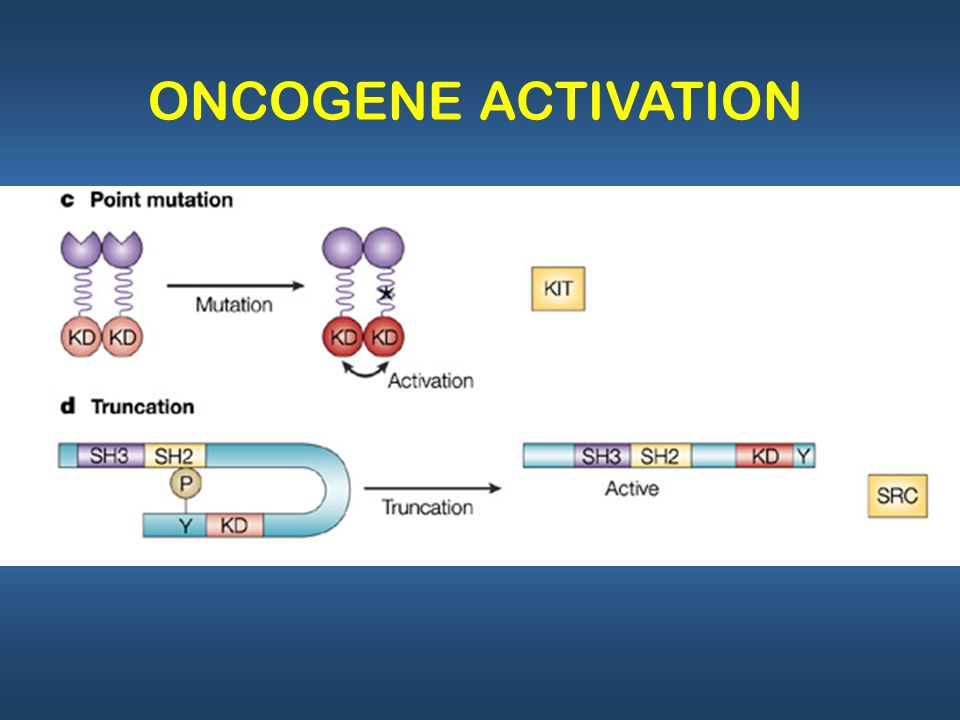
The Src-family kinases are examples of proteins that utilize autophosphorylation to sustain their activated states.[3] Src kinases are involved in intracellular signaling pathways that influence cell growth and cell adhesion strength. The latter contributes to the control of cell migration. In this way, src-kinase deregulation can enhance tumor growth and invasive potential of cancer cells.[2] The activity of src kinases is regulated by both phosphorylation and intramolecular interactions involving the SH2 and SH3 domains. The probable activation mechanism of src kinase in cancer is as follows:
- 1. The src kinase is kept in an inactive form through the binding of SH2 to a phosphotyrosine
- 2. Dephosphorylation of tyr-527 releases SH2 as well as SH3 domain.
- 3. Subsequent autophosphorylation of tyr-416 activates the kinase.
- 4. The constitutive activation of src kinase observed in cancer can be due to deletion of tyr-527, displacement of SH3 and SH2-mediated interactions by high affinity ligands with constantly autophosphorylated tyr-416.[2](Fig. 2).
Ataxia telangiectasia mutated kinase (ATM kinase)[edit]
ATM kinase, a member of the PI3-like family of serine/threonine kinases plays a critical role in maintaining the stability of the genome, which is of fundamental importance to the survival of all organisms. It exerts its effect by phosphorylating target proteins such as P53, MDM2 and chk2. Activation of ATM is facilitated by autophosphorylation. The inactive ATM exists as dimer, where the kinase domain of one monomer is bound to the internal domain of the other monomer, containing ser-1981. It will therefore be inaccessible to cellular substrates. In response to DNA damage, the kinase domain of one monomer phosphorylates ser-1981 of the other interacting ATM, resulting in subunit dissociation and ATM activation. The activated ATM triggers a sequence of events including cell cycle arrest which allows time for the repair of the damaged DNA. If damaged DNA is left unrepaired, it can lead to cell death or genomic instability, cancer and other pathologies.[23] Psp iso otome games.
See also[edit]
References[edit]
- ^Pecorino, L 2008, 'Molecular biology of cancer', Oxford University Press Inc., New York, U.S.A
- ^ abcFrame MC (Jun 2002). 'Src in cancer: deregulation and consequences for cell behaviour'. Biochimica et Biophysica Acta. 1602 (2): 114–30. doi:10.1016/s0304-419x(02)00040-9. PMID12020799.
- ^ abcdefghijklmPetsko, GA and Ringe, D 2009, 'Protein Structure and Function', Oxford University Press Inc., New York, U.S.A
- ^ abcSummers KC, Shen F, Sierra Potchanant EA, Phipps EA, Hickey RJ, Malkas LH (2011). 'Phosphorylation: the molecular switch of double-strand break repair'. International Journal of Proteomics. 2011: 373816. doi:10.1155/2011/373816. PMC3200257. PMID22084686.
- ^ abcSmith JA, Francis SH, Corbin JD (Nov 1993). 'Autophosphorylation: a salient feature of protein kinases'. Molecular and Cellular Biochemistry. 127–128: 51–70. doi:10.1007/BF01076757. PMID7935362.
- ^ abcdefghijkXu Q, Malecka KL, Fink L, Jordan EJ, Duffy E, Kolander S, Peterson JR, Dunbrack RL (Dec 2015). 'Identifying three-dimensional structures of autophosphorylation complexes in crystals of protein kinases'. Science Signaling. 8 (405): rs13. doi:10.1126/scisignal.aaa6711. PMC4766099. PMID26628682.
- ^Mol CD, Lim KB, Sridhar V, Zou H, Chien EY, Sang BC, Nowakowski J, Kassel DB, Cronin CN, McRee DE (Aug 2003). 'Structure of a c-kit product complex reveals the basis for kinase transactivation'. The Journal of Biological Chemistry. 278 (34): 31461–4. doi:10.1074/jbc.C300186200. PMID12824176.
- ^Meyers MJ, Pelc M, Kamtekar S, Day J, Poda GI, Hall MK, Michener ML, Reitz BA, Mathis KJ, Pierce BS, Parikh MD, Mischke DA, Long SA, Parlow JJ, Anderson DR, Thorarensen A (Mar 2010). 'Structure-based drug design enables conversion of a DFG-in binding CSF-1R kinase inhibitor to a DFG-out binding mode'. Bioorganic & Medicinal Chemistry Letters. 20 (5): 1543–7. doi:10.1016/j.bmcl.2010.01.078. PMID20137931.
- ^Bae JH, Lew ED, Yuzawa S, Tomé F, Lax I, Schlessinger J (Aug 2009). 'The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site'. Cell. 138 (3): 514–24. doi:10.1016/j.cell.2009.05.028. PMC4764080. PMID19665973.
- ^Huang Z, Chen H, Blais S, Neubert TA, Li X, Mohammadi M (Oct 2013). 'Structural mimicry of a-loop tyrosine phosphorylation by a pathogenic FGF receptor 3 mutation'. Structure. 21 (10): 1889–96. doi:10.1016/j.str.2013.07.017. PMC3839590. PMID23972473.
- ^Wu J, Li W, Craddock BP, Foreman KW, Mulvihill MJ, Ji QS, Miller WT, Hubbard SR (Jul 2008). 'Small-molecule inhibition and activation-loop trans-phosphorylation of the IGF1 receptor'. The EMBO Journal. 27 (14): 1985–94. doi:10.1038/emboj.2008.116. PMC2486273. PMID18566589.
- ^Nemecek C, Metz WA, Wentzler S, Ding FX, Venot C, Souaille C, Dagallier A, Maignan S, Guilloteau JP, Bernard F, Henry A, Grapinet S, Lesuisse D (Aug 2010). 'Design of potent IGF1-R inhibitors related to bis-azaindoles'. Chemical Biology & Drug Design. 76 (2): 100–6. doi:10.1111/j.1747-0285.2010.00991.x. PMID20545947.
- ^Jacobs MD, Caron PR, Hare BJ (Mar 2008). 'Classifying protein kinase structures guides use of ligand-selectivity profiles to predict inactive conformations: structure of lck/imatinib complex'. Proteins. 70 (4): 1451–60. doi:10.1002/prot.21633. PMID17910071.
- ^Wang J, Wu JW, Wang ZX (Dec 2011). 'Structural insights into the autoactivation mechanism of p21-activated protein kinase'. Structure. 19 (12): 1752–61. doi:10.1016/j.str.2011.10.013. PMID22153498.
- ^Staben ST, Feng JA, Lyle K, Belvin M, Boggs J, Burch JD, Chua CC, Cui H, DiPasquale AG, Friedman LS, Heise C, Koeppen H, Kotey A, Mintzer R, Oh A, Roberts DA, Rouge L, Rudolph J, Tam C, Wang W, Xiao Y, Young A, Zhang Y, Hoeflich KP (Feb 2014). 'Back pocket flexibility provides group II p21-activated kinase (PAK) selectivity for type I 1/2 kinase inhibitors'. Journal of Medicinal Chemistry. 57 (3): 1033–45. doi:10.1021/jm401768t. PMID24432870.
- ^Crawford JJ, Lee W, Aliagas I, Mathieu S, Hoeflich KP, Zhou W, Wang W, Rouge L, Murray L, La H, Liu N, Fan PW, Cheong J, Heise CE, Ramaswamy S, Mintzer R, Liu Y, Chao Q, Rudolph J (Jun 2015). 'Structure-Guided Design of Group I Selective p21-Activated Kinase Inhibitors'. Journal of Medicinal Chemistry. 58 (12): 5121–36. doi:10.1021/acs.jmedchem.5b00572. PMID26030457.
- ^Ndubaku CO, Crawford JJ, Drobnick J, Aliagas I, Campbell D, Dong P, Dornan LM, Duron S, Epler J, Gazzard L, Heise CE, Hoeflich KP, Jakubiak D, La H, Lee W, Lin B, Lyssikatos JP, Maksimoska J, Marmorstein R, Murray LJ, O'Brien T, Oh A, Ramaswamy S, Wang W, Zhao X, Zhong Y, Blackwood E, Rudolph J (Dec 2015). 'Design of Selective PAK1 Inhibitor G-5555: Improving Properties by Employing an Unorthodox Low-pK a Polar Moiety'. ACS Medicinal Chemistry Letters. 6 (12): 1241–6. doi:10.1021/acsmedchemlett.5b00398. PMC4677365. PMID26713112.
- ^Ferrao R, Zhou H, Shan Y, Liu Q, Li Q, Shaw DE, Li X, Wu H (Sep 2014). 'IRAK4 dimerization and trans-autophosphorylation are induced by Myddosome assembly'. Molecular Cell. 55 (6): 891–903. doi:10.1016/j.molcel.2014.08.006. PMC4169746. PMID25201411.
- ^Chao LH, Pellicena P, Deindl S, Barclay LA, Schulman H, Kuriyan J (Mar 2010). 'Intersubunit capture of regulatory segments is a component of cooperative CaMKII activation'. Nature Structural & Molecular Biology. 17 (3): 264–72. doi:10.1038/nsmb.1751. PMC2855215. PMID20139983.
- ^Rellos P, Pike AC, Niesen FH, Salah E, Lee WH, von Delft F, Knapp S (2010). 'Structure of the CaMKIIdelta/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation'. PLoS Biology. 8 (7): e1000426. doi:10.1371/journal.pbio.1000426. PMC2910593. PMID20668654.
- ^ abcBae JH, Schlessinger J (May 2010). 'Asymmetric tyrosine kinase arrangements in activation or autophosphorylation of receptor tyrosine kinases'. Molecules and Cells. 29 (5): 443–8. doi:10.1007/s10059-010-0080-5. PMID20432069.
- ^C O P E , Cytokines & Cells Online Pathfinder Encyclopedia, April 2012
- ^Bakkenist CJ, Kastan MB (Jan 2003). 'DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation'. Nature. 421 (6922): 499–506. doi:10.1038/nature01368. PMID12556884.